
The Krishnamurthy Lab
Publication
- Selection of Ribofuranose-isomer among Pentoses by Phosphorylation with Diamidophosphate. Cruz, H.A.; Krishnamurthy, R. Angew. Chemie Int. Ed. 2025 64, e202509810
Abstract
The unique structure-function relationship of the ribofuranose-ring in RNA has inspired various chemical pathways to select for the ribofuranose form (over the ribopyranose form), especially in a prebiotic context. Herein we show that the reaction of diamidophosphate (DAP) with the four pentoses - ribose, arabinose, xylose and lyxose - naturally selects the ribofuranose form more efficiently when compared to the other three pentoses. All four pentoses form the initial diamidophosphate-adduct – however, ribose undergoes rapid conversion to ribofuranose-1,2- and 2,3-cyclicphosphate products, while the other three pentoses show significant accumulation of the initially formed DAP-adducts and slow conversion to the cyclicphosphate products. Arabinose and xylose show a distribution between furanose-, and pyranose-1,2-cyclicphosphate products, while lyxose forms the furanose-1,2- and -pyranose-2,3-cyclicphosphate products. This trend is also manifested when starting from a mixture of pentoses where ribofuranose 1,2-amidocyclicphosphate dominates the product distribution. Such selection for ribofuranose among pentoses by phosphorylation (a) provides another venue of how a ribofuranose-structure can selectively emerge through abiotic chemical reaction suitable for elaboration towards RNA and (b) adds to the body of experimental work addressing the chemical origin of RNA.

- Phosphorylation of nucleosides by P-N bond species generated from prebiotic reduced phosphorus sources. Gull, M.; Cruz, H.A.; Krishnamurthy, R.; Pasek, M.A. Commun. Chem., 2025, 8:187

-
From Amino Acids to α-Keto Acids via β-Elimination and Transamination Initiates a Pathway to Prebiotic Reaction Networks. Ter-Ovanessian, L.MP.; Ryan, K.L.; Shaarda, J.; Stubbs, R.T.; Krishnamurthy, R.; Springsteen, G. Angew. Chem. Int. Ed. 2025, ASAP. https://doi.org/10.1002/anie.202507248
Abstract
α-Keto acids, such as pyruvate and glyoxylate, may have been critical in generating reaction networks at the origins of life due to their facile carbon-carbon bond formation and their hydrolytic stability. However, demonstrated prebiotic sources of these small α-keto acids have been limited by conditions required for their production which are not conducive for subsequent incorporation or transition into (proto)metabolic pathways. Here, we demonstrate an abiotic generation of α-keto acids from only two amino acids starting with the phosphorylation and dehydration of serine, coupled with a transamination with glycine, to produce both pyruvate and glyoxylate. This triggers an in-situ reaction pathway producing higher-order α-keto acids including amino acid precursors found in modern biology. These findings may help elucidate how protometabolic chemical networks can emerge on the early Earth under mild aqueous conditions leading to a coupled amino acid–α-keto acid chemical system capable of supporting a more robust metabolism.

- Abiotic aldol reactions of formaldehyde with ketoses and aldoses—Implications for the prebiotic synthesis of sugars by the formose reaction. Sutton, S.M.; Pulletikurti, S.; Lin, H.; Krishnamurthy, R.; Liotta, C. Chem, In Press, 2025. Chem 11, 102553, September 11, 2025

- Protocol for Preparing Cyclic-Phospholipid Decanoate and Glycerly-didecanoate-phosphate containing Vesicles. Veena, K.S.; Pulletikurti, S.; Deniz, A. A.; Krishnamurthy, R. STAR Protocols 5, 103283, 2024.
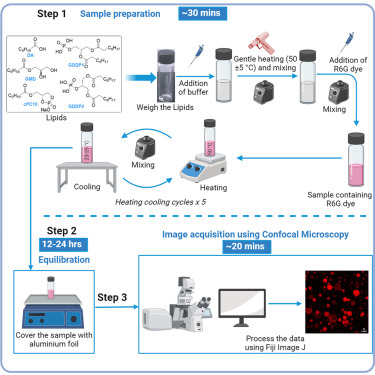
Cyclic-phospholipids-based vesicles can play a role in facilitating the chemical evolution of protocells from the structurally simple to the functionally more complex form. Here, we present a protocol for preparing decanoic acid-derived cyclic phospholipid and glyceryl-diester phosphate-containing vesicles. We describe steps for sample preparation, equilibration, and image acquisition using confocal microscopy. This protocol has the potential for preparing a wide variety of these phospholipid-based artificial cell constructs.
-
A Magnesium Binding Site And The Anomeric Effect Regulate The Abiotic Redox Chemistry Of Nicotinamide Nucleotides. Sebastianelli, L.; Kaur, H.; Chen, Z.; Krishnamurthy, R.; Mansy, S. S. Chem. Eur. J. 2024,30, e202400411) Open Access.

-
Experimentally modeling the emergence of prebiotically plausible phospholipid vesicles. Pulletikurti, S.; Veena, K. S.; Yadav, M.; Deniz, A. A.; Krishnamurthy, R. Chem. 2024 10, 1839-1867

- Cyclicphospholipids Enable a Protocellular Life Cycle. Toparlak, Ö.D.; Sebastianelli, L.; Ortuno, V.E.; Karki, M.; Xing, Y.; Szostak, J.; Krishnamurthy, R.; Mansy, S. ACS Nano 2023, 17, 23772–23783.

- Enhancing Prebiotic Phophorylation and Modulating the Regioselectivity of Nucleosides with Diamidophosphate. Cruz, H.A.; Jimenéz, E. I.; Krishnamurthy, R. J. Am. Chem. Soc. 2023, 43, 23781–23793.

-
Carbonyl Migration in Uronates Affords a Potential Prebiotic Pathway for Pentose Production. Yi, R.; Mojica, M.; Fahrenbach, A.; Cleaves, J.; Krishnamurthy, R.; Liotta, C. L. J. Am. Chem. Soc. Au. 2023, 3, 2522-2535.

- Microwave-Assisted One-Step Synthesis of 2',3'-cyclic Phosphates of Nucleosides. Veena, K.S.; Cruz, H. A.; Krishnamurthy, R. Current Protocols, 3, e834.2023 (doi: 10.1002/cpz1.834).

- The potential of glyoxylate as a prebiotic source molecule and a reactant in protometabolic pathways—The glyoxylose reaction. Krishamurthy, R.; Liotta, C. L. Chem. 9, 784-797, 2023. https://www.cell.com/chem/fulltext/S2451-9294(23)00126-2

Summary
Recent work has demonstrated glyoxylate as a versatile source molecule and reactant under plausible prebiotic conditions, giving rise to protometabolic analogs of the r-TCA cycle while enabling the synthesis of hydroxy acids, α-amino acids, and orotate. These observations suggest that glyoxylate, as a “carboxylated-formaldehyde” equivalent, has the potential to play an even greater role in prebiotic chemistry, providing an alternative to formaldehyde-based reactions. Thus, and inspired by the Eschenmoser’s glyoxylate scenario, we propose a distinct “glyoxylose reaction” framework where the initial formation of glycolaldehyde from glyoxylate is followed by a series of repetitive (1) aldol condensations with glyoxylate, (2) carbonyl migrations, and (3) decarboxylations. This alternative to the formose reaction is hypothesized to provide linear sugars and sugar acids while harboring the potential for autocatalytic pathways. A demonstration of this chemistry would expand the role of glyoxylate as a versatile molecule in prebiotic chemistry and stimulate further the search for its prebiotic provenance. - Prebiotic Chemistry of Phosphite: Mild Thermal Routes to Form Condensed-P Energy Currency Molecules Leading up to the Formation of Organophosphorus Compounds. Gull, M.; Fent, T.; Cruz, H. A.; Krishnamurthy, R.; Pasek, M. Life, 13, 920 2023. https://doi.org/10.3390/life13040920


- Erythrose and Threose: Carbonyl Migrations, Epimerizations, Aldo and Oxidative Fragmentation Reactions Under Plausible Prebiotic Conditions. Yi, R.; Kern, R.; Pollet, P.; Lin, H.; Krishnamurthy, R.; Liotta, C. L. Chem. Eur. J. 2022. https://doi.org/10.1002/chem.202202816

- One-pot chemical pyro- and tri-phosphorylation of peptides using diamidophosphate in water. Lin, H.; Leman, L. J.; Krishnamurthy, R. Chem. Sci. 2022, 13, 13741-13747.


- A Plausible Prebiotic Path to Nucleosides: Ribosides and Related Aldosides are Generated from Ribulose, Fructose and Similar Abiotic Precursors. Roche, T, P.; Fialho, D. M.; Menor-Salvan, C.; Krishnamurthy, R.; Schuster, G. B.; Hud, N. V. Chem. Eur. J. 2022. https://doi.org/10.1002/chem.202203036

- Nucleobases in Meteorites to Nucleobases in RNA and DNA? Krishnamurthy, R.; Goldman, A.D.; Liberles, D. A.; Rogers, K. L.; Tor, Y. J. Mol. Evol. 2022, 90, pages 328–331.

- Prebiotic Synthesis of α-amino acids orotate from α-ketoacids potentiates a transition to extant metabolic pathways. Pulletikurti, S.; Yadav, M.; Springsteen, G.; Krishnamurthy, R. Nat. Chem. 2022, 14, 1142–1150.


- Frontiers in Prebiotic Chemistry and Early Earth Environments. Müller, U.F.; Elsila, J.; Trail, D.; DasGupta, S.; Giese, C-C.; Watlon, C. R.; Cohen, Z. R.; Stolar, T.; Krishnamurthy, R.; Lyons, T.; Rogers, K. L.; Williams, L.D. Orig. Life Evol. Biosph. 2022, 90, 328–331.

- Synthesis and Hydrolytic Stability of Cyclic Phosphatidic Acids: Implications for Synthetic- and Proto-cell Studies. Egas, V. O; Pulletikurti, S.; Kollery, V. S.; Krishnamurthy. Chem. Commun. 2022, 58, 6231-6234. This article is part of the themed collection: 2021 CRSI Medal Winners Collection

- Cyanide as a primordial reductant enables a protometabolic reductive glyoxylate pathway. Yadav, M.; Pulletikurti, S.; Yerabolu, J. R.; Krishnamurthy, R. Nat. Chem. 2022, 14, 170-178.
Abstract
Investigation of prebiotic metabolic pathways is predominantly based on abiotically replicating the reductive citric acid cycle. While attractive from a parsimony point of view, attempts using metal/mineral-mediated reductions have produced complex mixtures with inefficient and uncontrolled reactions. Here we show that cyanide acts as a mild and efficient reducing agent mediating abiotic transformations of tricarboxylic acid intermediates and derivatives. The hydrolysis of the cyanide adducts followed by their decarboxylation enables the reduction of oxaloacetate to malate and of fumarate to succinate, whereas pyruvate and α-ketoglutarate themselves are not reduced. In the presence of glyoxylate, malonate and malononitrile, alternative pathways emerge that bypass the challenging reductive carboxylation steps to produce metabolic intermediates and compounds found in meteorites. These results suggest a simpler prebiotic forerunner of today’s metabolism, involving a reductive glyoxylate pathway without oxaloacetate and α-ketoglutarate—implying that the extant metabolic reductive carboxylation chemistries are an evolutionary invention mediated by complex metalloproteins.

- A Plausible Prebiotic One-Pot Synthesis of Orotate and Pyruvate Suggestive of Common Protometabolic Pathways. Clay, A. P.; Cooke, R. E.; Kumar, R.; Yadav, M.; Krishnamurthy, R.; Springsteen, G. Angew. Chemie. Int. Ed. 2022, e202112572.


-
Concurrent Prebiotic Formation of Nucleoside-Amidophosphates and Nucleoside-Triphosphates Potentiates Transition from Abiotic to Biotic Polymerization. Lin, H.; Jiménez, E. I.; Arriola, J. T.; Müller, U.; Krishnamurthy, R. Angew. Chemie. Int. Ed. 2022, 61, e202113625.


-
Depsipeptide Nucleic Acids: Prebiotic Formation, Oligomerization, and Self-Assembly of a New Proto-Nucleic Acid Candidate. Fialho, D. M.; Karunakaran, S. C.; Greeson, K. W.; Martinéz, I.; Schuster, G. B.; Krishnamurthy, R.; Hud, V. J. Am. Chem. Soc. 2021, 143, 13525-13537.


- Towards an understanding of the molecular mechanisms of variable unnatural base pair behavior—A biophysical analysis of dNaM-dTPT3. Karadeema, R. J.; Morris, S. E., Lairson, L. L.; Krishnamurthy, R. Chem. Eur. J. 2021, 27, 13991-13997.



-
Separations of Carbohydrates with Noncovalent Shift Reagents by Frequency-Modulated Ion Mobility-Orbitrap Mass Spectrometry. McKenna, K. R.; Clowers, B. H.; Krishnamurthy, R.; Liotta, C. L.; Fernández, F. M. J. Am. Soc. Mass Spectrom. 2021, 32, 9, 2472–2480


- Diamidophosphate (DAP) – A Plausible Prebiotic Phosphorylating Reagent with a Chem to BioChem Potential? Osumah, A.; Krishnamurthy, R. ChemBioChem, 2021, 22, 3001-3009. (Open Access)


- Transcriptional processing of an unnatural base pair by eukaryotic RNA polymerase II. Oh. J.; Shin, J.; Unarta, C. I.; Wang, W.; Feldman, A. W.; Karadeema, R. J.; Xu, L.; Xu, J.; Chong, J.; Krishnamurthy, R.; Huang, X.; Romesberg, F. E.; Wang, D. Nat. Chem. Biol. 2021, 17, 906-914

- Prebiotic Phosphorylation and Concomitant Oligomerization of Deoxynucleosides to form DNA. Jiménez, E. I.; Gibard, C.; Krishnamurthy, R. Angew. Chemie. Int. Ed. 2021, 60, 10775-10783

- Noncovalent Helicene Structure between Nucleic Acids and Cyanuric Acid. Alenaizan, A.; Fauche, K.; Krishnamurthy, R.; Sherril., D. C. Chem. Eur. J. 2021, 27, 4043-4052.


- The Unexpected Base‐pairing Behavior of Cyanuric Acid in RNA and Ribose versus Cyanuric Acid Induced Helicene Assembly of Nucleic Acids: Implications for the pre‐RNA Paradigm. Anderson, B.; Fauche, K.; Karunakaran, S.; Yerabolu, J.; Hud, N.; Krishnamurthy, R. Chem. Eur. J. 2021, 27, 4033-4042.


- Prebiotically plausible RNA activation compatible with ribozyme-catalyzed ligation. Song, E. Y.; Jiménez, E. I.; Lin, H.; Lay, K. V.; Krishnamurthy, R.; Mutschler, H. Angew. Chemie. 2020, 60, 2952-2957.


- Systems Chemistry in the Chemical Origins of Life: The 18th Camel Paradigm. Krishnamurthy, R. J. Systems Chem. 2020, 8, 40-62 (Special Issue Dedicated to Günter von Kiedrowski). OPEN ACCESS

- A Plausible Metal-Free Ancestral Analogue of the Krebs Cycle Composed Entirely of alpha-ketoacids. Stubbs, R.T.; Yadav, M.; Krishnamurthy, R.; Springsteen, G. Nat. Chem. 2020. 12, 1016-1022.


- Organic Acid Shift Reagents for the Discrimination of Carbohydrate Isobars by Ion Mobility-Mass Spectrometry. McKenna, K. R.; Krishnamurthy, R.; Liotta, C.; Fernandez, F. Analyst, 2020 145, 8008-8015


- A Sensitive Quantitative Analysis of Abiotically Synthesized Short Homopeptides using Ultraperformance Liquid Chromatography and Time-of-Flight Mass Spectrometry. Parker, E. T.; Karki, M.; Glavin, D. P.; Dworkin, J. P.; Krishnamurthy, R. J. Chrom. A 2020, 1630, 461509


- Mutually stabilzing interactions between proto-peptides and RNA. Frenkel-Pinter, M.; Hyanes, J. W.; Moheyeldin, A. M.; Martinc C, Sargon, A. B.; Petrov, A. S.; Krishnamurthy, R.; Hud, N.; Williams, L. D.; Leman, L.J. Nat. Commun. 2020, 11, article no. 3137.


- Introduction: Chemical Evolution and the Origins of Life. Krishnamurthy, R.; Hud, N. Chem. Rev. 2020, 120, 11, 4613–4615

- New codons for efficient production of unnatural proteins in a semisynthetic organism. Fischer, E.C.; Hashimoto, K.; Zhang, Y.; Feldman, A.W.; Dient, V.T.; Karadeema, R.J.; Adhikary, R.; Ledbetter, M.P.; Krishnamurthy, R.; Romesberg, F.L. Nat. Chem. Biol. 2020 16, 570–576(2020)
 Natural organisms use a four-letter genetic alphabet that makes available 64 triplet codons, of which 61 are sense codons used to encode proteins with the 20 canonical amino acids. We have shown that the unnatural nucleotides dNaM and dTPT3 can pair to form an unnatural base pair (UBP) and allow for the creation of semisynthetic organisms (SSOs) with additional sense codons. Here, we report a systematic analysis of the unnatural codons. We identify nine unnatural codons that can produce unnatural protein with nearly complete incorporation of an encoded noncanonical amino acid (ncAA). We also show that at least three of the codons are orthogonal and can be simultaneously decoded in the SSO, affording the first 67-codon organism. The ability to incorporate multiple, different ncAAs site specifically into a protein should now allow the development of proteins with novel activities, and possibly even SSOs with new forms and functions.
Natural organisms use a four-letter genetic alphabet that makes available 64 triplet codons, of which 61 are sense codons used to encode proteins with the 20 canonical amino acids. We have shown that the unnatural nucleotides dNaM and dTPT3 can pair to form an unnatural base pair (UBP) and allow for the creation of semisynthetic organisms (SSOs) with additional sense codons. Here, we report a systematic analysis of the unnatural codons. We identify nine unnatural codons that can produce unnatural protein with nearly complete incorporation of an encoded noncanonical amino acid (ncAA). We also show that at least three of the codons are orthogonal and can be simultaneously decoded in the SSO, affording the first 67-codon organism. The ability to incorporate multiple, different ncAAs site specifically into a protein should now allow the development of proteins with novel activities, and possibly even SSOs with new forms and functions. - Chemical Origins of Life: Its Engagement with Society. Krishnamurthy, R. Trends in Chemistry, 2020, 2, 406-409. Read Accepted Version

- Nanopore Sequencing of an Expanded Genetic Alphabet RevealsHigh-Fidelity Replication of a Predominantly HydrophobicUnnatural Base Pair. Ledbetter, M.P.; Craig, J.M.; Karadeema, R.J.; Noakes, M.T.; Kim, H.C.; Abell, S.J.; Huang, J.R.; Anderson, B.A.; Krishnamurthy, R.; Gundlach, J. H.; Romesberg, F.E. J. Am. Chem. Soc. 2020, 142, 2110-2114.
 ABSTRACT:Unnatural base pairs (UBPs) have been developed and used for a variety ofin vitroapplications as well as for theengineering of semisynthetic organisms (SSOs) that store and retrieve increased information. However, these applications arelimited by the availability of methods to rapidly and accurately determine the sequence of unnatural DNA. Here we report thedevelopment and application of the MspA nanopore to sequence DNA containing the dTPT3−dNaMUBP. Analysis of twosequence contexts reveals that DNA containing the UBP is replicated with an efficiency andfidelity similar to that of natural DNAand sufficient for use as the basis of an SSO that produces proteins with noncanonical amino acids.
ABSTRACT:Unnatural base pairs (UBPs) have been developed and used for a variety ofin vitroapplications as well as for theengineering of semisynthetic organisms (SSOs) that store and retrieve increased information. However, these applications arelimited by the availability of methods to rapidly and accurately determine the sequence of unnatural DNA. Here we report thedevelopment and application of the MspA nanopore to sequence DNA containing the dTPT3−dNaMUBP. Analysis of twosequence contexts reveals that DNA containing the UBP is replicated with an efficiency andfidelity similar to that of natural DNAand sufficient for use as the basis of an SSO that produces proteins with noncanonical amino acids. - Chemistry of Abiotic Nucleotide Synthesis. Yadav, M.; Kumar, R.; Krishnamurthy, R. Chem. Rev. 2020.120, 11, 4766–4805 (open access)

Abstract
The chemistry of abiotic nucleotide synthesis of RNA and DNA in the context of their prebiotic origins on early earth is a continuing challenge. How did (or how can) the nucleotides form and assemble from the small molecule inventories and under conditions that prevailed on early earth 3.5–4 billion years ago? This review provides a background and up-to-date progress that will allow the reader to judge where the field stands currently and what remains to be achieved. We start with a brief primer on the biological synthesis of nucleotides, followed by an extensive focus on the prebiotic formation of the components of nucleotides—either via the synthesis of ribose and the canonical nucleobases and then joining them together or by building both the conjoined sugar and nucleobase, part-by-part—toward the ultimate goal of forming RNA and DNA by polymerization. The review will emphasize that there are—and will continue to be—many more questions than answers from the synthetic, mechanistic, and analytical perspectives. We wrap up the review with a cautionary note in this context about coming to conclusions as to whether the problem of chemistry of prebiotic nucleotide synthesis has been solved.
- The Oligomerization of Glucose Under Plausible Prebiotic Conditions. Li, Z.; Li, L.; McKenna, K. R.; Pollet, P.; Gelbaum, L.; Fernandez, F.; Krishnamurthy, R.; Liotta, C. L. Orig. Life Evol. Biosph. 2019, 49, 225–240.

Abstract
The prebiotic origin of polysaccharides, the largest class of biopolymers by mass in extant biology, has seldom been investigated experimentally. Herein, we report on the acid-catalyzed condensation of aqueous solutions of glucose, a model monosaccharide, under plausible prebiotic conditions employing a wet-dry (night-day) protocol with 0.01 M HCl at 50 °C. This protocol leads to the formation of oligosaccharides containing up to eight monomeric units identified by high resolution mass spectrometry. The regio- and stereochemistry of the oligomeric acetal linkages, as well as the quantitative analysis of glucose conversion, are elucidated by combining 1H, 13C and 2D NMR spectroscopy. Ten out of eleven possible acetal linkages, including α- and β- anomers, have been identified with the α- and β- 1,6-acetals being the dominant linkages observed. In addition, the acid-catalyzed oligomerization of several glucose disaccharides such as cellobiose, maltose, and gentiobiose are presented along with an accompanying comparison with the corresponding oligomerization of glucose.
- Carbohydrate Isomer Resolution via Multi-site Derivatization Cyclic Ion Mobility-Mass Spectrometry. McKenna, K.R.; Li, L.; Baker, A.; Krishnamurthy, R.; Liotta, C.L.; Fernandez, F. Analyst, 2019,144, 7220-7226.
 Abstract
AbstractOligosaccharides serve many roles in extant life and may have had a significant role in prebiotic chemistry on the early Earth. In both these contexts, the structural and isomeric diversity among carbohydrates presents analytical challenges necessitating improved separations. Here, we showcase a chemical derivatization approach, where 3-carboxy-5-nitrophenylboronic acid (3C5NBA) is used to label vicinal hydroxyl groups, amplifying the structural difference between isomers. We explore the applicability of state-of-the-art ion mobility – mass spectrometry (IM-MS) instrumentation in the analysis of derivatized carbohydrates. In particular we focus on the resolving power required for IM separation of derivatized isomers. A recently developed cyclic ion mobility (cIM) mass spectrometer (MS) was chosen for this study as it allows for multi-pass IM separations, with variable resolving power (Rp). Three passes around the cIM (Rp ∼ 120) enabled separation of all possible pairs of four monosaccharide standards, and all but two pairs of eight disaccharide standards. Combining cIM methodology with tandem mass spectrometry (MS/MS) experiments allowed for the major products of each of the 3C5NBA carbohydrate derivatization reactions to be resolved and unequivocally identified.
- Synthesis of 2-thioorotidine and comparison of its unusual instability with its canonical pyrimidine counterparts. Kumar, R.; Springsteen, G.; Krishnamurthy, R. J. Org. Chem. 2019, 84, 14427-14435.

Abstract
2-Thiopyrimidine nucleosides exhibit properties that are interesting from both a biological/medicinal and origins of life chemistry point of view. We report here the first synthesis of the nucleoside 2-thioorotidine and our observations on its unexpected lability with respect to its N-glycosidic bond when compared with its corresponding canonical pyrimidine counterparts. We hypothesize that the cause of the lability of the N-glycosidic bond is due to the combined steric and electronic effects from the 2-thio- and the 6-carboxyl groups, a lability that could, in turn, be useful for further chemical transformations.
- Prebiotic Phosphorylation of Uridine using Diamidophosphate in Aerosols. Catsañeda, A.D.; Li, Z.; Joo, T.; Benham, K.; Burcar, B.T.; Krishnamurthy, R.; Liotta, C.L.; Ng, N.L.; Orlando, T.M. Scientific Reports volume 9, Article number: 13527 (2019).

Abstract
One of the most challenging fundamental problems in establishing prebiotically plausible routes for phosphorylation reactions using phosphate is that they are thermodynamically unfavorable in aqueous conditions. Diamidophosphate (DAP), a potentially prebiotically relevant compound, was shown to phosphorylate nucleosides in aqueous medium, albeit at a very slow rate (days/weeks). Here, we demonstrate that performing these reactions within an aerosol environment, a suitable model for the early Earth ocean-air interface, yields higher reaction rates when compared to bulk solution, thus overcoming these rate limitations. As a proof-of-concept, we demonstrate the effective conversion (~6.5–10%) of uridine to uridine-2′,3′-cyclophosphate in less than 1 h. These results suggest that aerosol environments are a possible scenario in which prebiotic phosphorylation could have occurred despite unfavorable rates in bulk solution.
- The role of sugar-backbone heterogeneity and chimeras in the simultaneous emergence of RNA and DNA. Bhowmik, S.; Krishnamurthy, R. Nature Chemistry, 2019, 11, 1009-1018. Read the article online
 The emergence of pristine RNA and DNA on the early Earth would have been hindered by a lack of specificity in their prebiotic syntheses. Now, it has been shown that chimeric sequences—with a mixture of RNA and DNA backbones—mediate the template-directed ligation of oligomers present in mixtures of nucleic acids, enabling the simultaneous appearance of RNA and DNA.
The emergence of pristine RNA and DNA on the early Earth would have been hindered by a lack of specificity in their prebiotic syntheses. Now, it has been shown that chimeric sequences—with a mixture of RNA and DNA backbones—mediate the template-directed ligation of oligomers present in mixtures of nucleic acids, enabling the simultaneous appearance of RNA and DNA. - Cyclophospholipids Increase Protocellular Stability to Metal Ions. Toparlak, D. O.; Karki, M.; Egas Ortuno, V.; Krishnamurthy, R.; Mansy, S. S. Small, 2019, 1903381, 1-8

Special Issue:Artificial Biology–Molecular Design and Cell Mimicry
Selected for Hot Topics: Vesicles - Bis(dimethylamino)phosphoridiamidate: A Reagent for the Regioselective Cyclophosphorylation of cis-Diols Enbabling One-Step Access to High-Value Target Cyclophosphates. Yadav, M.; Krishnamurthy, R. Org. Lett. 2019, 21, 7400-7404.


- Selective Incorporation of Proteinaceous over Nonproteinaceous Cationic Amino Acids in Model Prebiotic Oligomerization Reactions.
 Abstract
AbstractNumerous long-standing questions in origins-of-life research center on the history of biopolymers. For example, how and why did nature select the polypeptide backbone and proteinaceous side chains? Depsipeptides, containing both ester and amide linkages, have been proposed as ancestors of polypeptides. In this paper, we investigate cationic depsipeptides that form under mild dry-down reactions. We compare the oligomerization of various cationic amino acids, including the cationic proteinaceous amino acids (lysine, Lys; arginine, Arg; and histidine, His), along with nonproteinaceous analogs of Lys harboring fewer methylene groups in their side chains. These analogs, which have been discussed as potential prebiotic alternatives to Lys, are ornithine, 2,4-diaminobutyric acid, and 2,3-diaminopropionic acid (Orn, Dab, and Dpr). We observe that the proteinaceous amino acids condense more extensively than these nonproteinaceous amino acids. Orn and Dab readily cyclize into lactams, while Dab and Dpr condense less efficiently. Furthermore, the proteinaceous amino acids exhibit more selective oligomerization through their α-amines relative to their side-chain groups. This selectivity results in predominantly linear depsipeptides in which the amino acids are α-amine−linked, analogous to today’s proteins. These results suggest a chemical basis for the selection of Lys, Arg, and His over other cationic amino acids for incorporation into proto-proteins on the early Earth. Given that electrostatics are key elements of protein−RNA and protein−DNA interactions in extant life, we hypothesize that cationic side chains incorporated into proto-peptides, as reported in this study, served in a variety of functions with ancestral nucleic acid polymers in the early stages of life.
- Optimization of Replication, Transcription, and Translation in a Semi-Synthetic Organism. Feldman, A. W.; Dien, V. T.; Karadeema, R. J.; Fischer, E. C.; Y, Y.; Anderson, B.; Krishnamurthy, R.; Chen, J. S.; L, L.; Romesberg, F. E. J. Am. Chem. Soc. 2019, 141, 10644-10653.
Abstract

Previously, we reported the creation of a semi-synthetic organism (SSO) that stores and retrieves increased information by virtue of stably maintaining an unnatural base pair (UBP) in its DNA, transcribing the corresponding unnatural nucleotides into the codons and anticodons of mRNAs and tRNAs, and then using them to produce proteins containing noncanonical amino acids (ncAAs). Here we report a systematic extension of the effort to optimize the SSO by exploring a variety of deoxy- and ribonucleotide analogues. Importantly, this includes the first in vivo structure–activity relationship (SAR) analysis of unnatural ribonucleoside triphosphates. Similarities and differences between how DNA and RNA polymerases recognize the unnatural nucleotides were observed, and remarkably, we found that a wide variety of unnatural ribonucleotides can be efficiently transcribed into RNA and then productively and selectively paired at the ribosome to mediate the synthesis of proteins with ncAAs. The results extend previous studies, demonstrating that nucleotides bearing no significant structural or functional homology to the natural nucleotides can be efficiently and selectively paired during replication, to include each step of the entire process of information storage and retrieval. From a practical perspective, the results identify the most optimal UBP for replication and transcription, as well as the most optimal unnatural ribonucleoside triphosphates for transcription and translation. The optimized SSO is now, for the first time, able to efficiently produce proteins containing multiple, proximal ncAAs.
- Geochemical Sources and Availability of Amidophosphates on the Early Earth. Gibard, C.; Jiménez, E. I.; Gorrell, I. B.; Kee, T. P.; Pasek, M. A.; Krishnamurthy, R. Angew. Chemie Int. Ed. 2019, 58, 8151-8155.

Abstract
Phosphorylation of (pre)biotically relevant molecules in aqueous medium has recently been demonstrated using water‐soluble diamidophosphate (DAP). Questions arise relating to the prebiotic availability of DAP and other amidophosphosphorus species on the early earth. Herein, we demonstrate that DAP and other amino‐derivatives of phosphates/phosphite are generated when Fe3P (proxy for mineral schreibersite), condensed phosphates, and reduced oxidation state phosphorus compounds, which could have been available on early earth, are exposed to aqueous ammonia solutions. DAP is shown to remain in aqueous solution under conditions where phosphate is precipitated out by divalent metals. These results show that nitrogenated analogues of phosphate and reduced phosphite species can be produced and remain in solution, overcoming the thermodynamic barrier for phosphorylation in water, increasing the possibility that abiotic phosphorylation reactions occurred in aqueous environments on early earth.
- Prebiotic phosphorylation of 2-thiouridine provides either nucleotides or DNA building blocks via photoreduction. Xu, J.; Green, N. J.; Gibard, C.; Krishnamurthy, R.; Sutherland, J. D. Nat. Chem. 2019,11, 457-462. view the paper online

- Experimentally Investigating the Origin of DNA/RNA on Early Earth. (Comment) Krishnamurthy, R. Nature Communications, 2018, 9:5175 DOI 10.1038/s41467-018-07212-y

Abstract
There are varied views about how the molecules of life may have appeared on early Earth. Nowhere is this divergence more acute than in the origins of DNA/RNA and has become a matter of constant deliberations.
-
Base-Mediated Cascade Aldol Addition and Fragmentation Reactions of Dihydroxyfumaric Acid and Aromatic Aldehydes: Controlling Chemodivergence via Choice of Base, Solvent, and Substituents. Ward, G.; Liotta, C. L.; Krishnamurthy, R.; France, S. J. Org. Chem. 2018, 83, 14219-14233. Featured Article.

- Chimeric XNA - An Unconventional Design for Orthogonal Informational Systems. Efthymiou, T.; Gavette, J.; Stoop. M.; De Riccardis, F.; Froyen, M.; Herdewijn, P.; Krishnamurthy, R. Chem. Eur. J. 2018, 24, 12811-12819.
 Greater than the sum of its parts: The combination of RNA (DNA) units with non‐base pairing or weakly base pairing XNA residues (such as ribuloNA and isoGNA) in an alternating repeat pattern, gives rise to dimeric and trimeric orthogonal oligonucleotides capable of duplex formation with unusual properties. This approach suggests an unconventional design paradigm for generating novel orthogonal chimeric nucleic acid informational systems in which both the backbone composition and nucleobase sequence can encode for information.
Greater than the sum of its parts: The combination of RNA (DNA) units with non‐base pairing or weakly base pairing XNA residues (such as ribuloNA and isoGNA) in an alternating repeat pattern, gives rise to dimeric and trimeric orthogonal oligonucleotides capable of duplex formation with unusual properties. This approach suggests an unconventional design paradigm for generating novel orthogonal chimeric nucleic acid informational systems in which both the backbone composition and nucleobase sequence can encode for information. 
- Life's Biological Chemistry: A Destiny or Destination Starting from Prebiotic Chemistry? Krishnamurthy, R. Chem. Eur. J. 2018, 24, 16708-16715.
 Research into understanding the origins—and evolution—of life has long been dominated by the concept of taking clues from extant biology and extrapolating its molecules and pathways backwards in time. This approach has also guided the search for solutions to the problem of how contemporary biomolecules would have arisen directly from prebiotic chemistry on early earth. However, the continuing difficulties in finding universally convincing solutions in connecting prebiotic chemistry to biological chemistry should give us pause, and prompt us to rethink this concept of treating extant life's chemical processes as the sole end goal and, therefore, focusing only, and implicitly, on the respective extant chemical building blocks. Rather, it may be worthwhile “to set aside the goal” and begin with what would have been plausible prebiotic reaction mixtures (which may have no obvious or direct connection to life's chemical building blocks and processes) and allow their chemistries and interactions, under different geochemical constraints, to guide and illuminate as to what processes and systems can emerge. Such a conceptual approach gives rise to the prospect that chemistry of life‐as‐we‐know‐it is not the only result (not a “destiny”), but one that has emerged among many potential possibilities (a “destination”). This postulate, in turn, could impact the way we think about chemical signatures and criteria used in the search for alternative and extraterrestrial “life”. As a bonus, we may discover the chemistries and pathways naturally that led to the emergence of life as we know it.
Research into understanding the origins—and evolution—of life has long been dominated by the concept of taking clues from extant biology and extrapolating its molecules and pathways backwards in time. This approach has also guided the search for solutions to the problem of how contemporary biomolecules would have arisen directly from prebiotic chemistry on early earth. However, the continuing difficulties in finding universally convincing solutions in connecting prebiotic chemistry to biological chemistry should give us pause, and prompt us to rethink this concept of treating extant life's chemical processes as the sole end goal and, therefore, focusing only, and implicitly, on the respective extant chemical building blocks. Rather, it may be worthwhile “to set aside the goal” and begin with what would have been plausible prebiotic reaction mixtures (which may have no obvious or direct connection to life's chemical building blocks and processes) and allow their chemistries and interactions, under different geochemical constraints, to guide and illuminate as to what processes and systems can emerge. Such a conceptual approach gives rise to the prospect that chemistry of life‐as‐we‐know‐it is not the only result (not a “destiny”), but one that has emerged among many potential possibilities (a “destination”). This postulate, in turn, could impact the way we think about chemical signatures and criteria used in the search for alternative and extraterrestrial “life”. As a bonus, we may discover the chemistries and pathways naturally that led to the emergence of life as we know it. - Heterogeneous Pyrophosphate Linked DNA-Oligonucleotides: Aversion for DNA but Affinity for RNA. Anderson, B.; Krishnamurthy, R. Chem. Eur. J. 2018, 24, 6837-6842.
 Growing a backbone: A systematic study of oligonucleotides with increasing incorporation of the pyrophosphate‐linked DNA unit reveals a destabilizing effect for DNA–DNA, but not RNA–DNA duplexes.
Growing a backbone: A systematic study of oligonucleotides with increasing incorporation of the pyrophosphate‐linked DNA unit reveals a destabilizing effect for DNA–DNA, but not RNA–DNA duplexes. - Effect of temperature modulations on TEMPO-mediated regioselective oxidation of unprotected carbohydrates and nucleosides. Yadav, M.; Liotta, C. L.; Krishnamurthy, R. Biorg. Med. Chem. Lett. 2018, 28, 2759-2765.
 Regioselective oxidation of unprotected and partially protected oligosaccharides is a much sought-after goal. Herein, we report a notable improvement in the efficiency of TEMPO-catalyzed oxidation by modulating the temperature of the reaction. Mono-, di-, and tri-saccharides are oxidized regioselectively in yields of 75 to 92%. The present method is simple to implement and is also applicable for selective oxidations of other mono- and poly-hydroxy compounds including unprotected and partially protected nucleosides.
Regioselective oxidation of unprotected and partially protected oligosaccharides is a much sought-after goal. Herein, we report a notable improvement in the efficiency of TEMPO-catalyzed oxidation by modulating the temperature of the reaction. Mono-, di-, and tri-saccharides are oxidized regioselectively in yields of 75 to 92%. The present method is simple to implement and is also applicable for selective oxidations of other mono- and poly-hydroxy compounds including unprotected and partially protected nucleosides. - Rapid Resolution of Carbohydrate Isomers via Multi-site Derivatization Ion Mobility-Mass Spectrometry. Li, L,; McKenna, K. R.; Li, Z.; Yadav, M.; Krishnamurthy, R.; Liotta, C. L.; Facundo, M. F. Analyst, 2018,143, 949-955.
 Identifying small sugar isomers can be challenging by ion mobility-mass spectrometry (IM-MS) alone due to their small collision cross section differences.
Identifying small sugar isomers can be challenging by ion mobility-mass spectrometry (IM-MS) alone due to their small collision cross section differences. - Linked cycles of oxidative decarboxylation of glyoxylate as protometabolic analgos of the citric acid cycle. Springsteen, G.; Yerabolu, J. R.; Nelos, J.; Rhea, C. J.; Krishnamurthy, R. Nature Communications, 2018, 9:91; DOI: 10.1038/s41467-017-0259-0
Abstract:
The development of metabolic approaches towards understanding the origins of life, which have focused mainly on the citric acid (TCA) cycle, have languished—primarily due to a lack of experimentally demonstrable and sustainable cycle(s) of reactions. We show here the existence of a protometabolic analog of the TCA involving two linked cycles, which convert glyoxylate into CO2 and produce aspartic acid in the presence of ammonia. The reactions proceed from either pyruvate, oxaloacetate or malonate in the presence of glyoxylate as the carbon source and hydrogen peroxide as the oxidant under neutral aqueous conditions and at mild temperatures. The reaction pathway demonstrates turnover under controlled conditions. These results indicate that simpler versions of metabolic cycles could have emerged under potential prebiotic conditions, laying the foundation for the appearance of more sophisticated metabolic pathways once control by (polymeric) catalysts became available.
- Glycosylation of a model proto-RNA nucleobase with non-ribose sugars: Implications for the prebiotic synthesis of nucleosides. Filaho, D.; Clarke, K.; Moore, M.; Schuster, G.; Krishnamurthy, R.; Hud, N. Org. Biomol. Chem. 2018, 16, 1263-1271

- Phosphorylation, oligomerization and self-assembly in water under potential prebiotic conditions. Gibard, C.; Bhowmik, S.; Karki, M.; Kim, E.-K.; Krishnamurthy, R. Nat. Chem. 2018, 10, 212-217.


- Elongation of Model Prebiotic Proto-peptides by Continious Monomer Feeding. Yu, S-S.; Martin, S.; Blanchard, M.; Soper-Hopper, M.; Krishnamurthy, R.; Fernandez, F.; Hud, N,; Schork, F. J.; Gover, M. Macromolecules, 2017, 50, 9286–9294.

- Surveying the sequence diversity of model prebiotic peptides by mass spectrometry. Forsythe, J.G., Petrov, A. S.; Sheng-Sheng, Y., Krishnamurthy, R., Grover, M., Hud, N. V., Facundo, M. F. Proc. Natl. Acad. Soc. 2017, 114, E7652-E7659.

- Nitrogenous Derivatives of Phosphorus and the Origins of Life: Plausible Prebiotic Phosphorylating Agents in Water. Karki, M.; Gibard, C.; Bhowmik, S.; Krishnamurthy, R. Life, 2017, 7, 32.


- Orotidine Containing RNA: Implications for the Hierarchical Selection (Systems Chemistry Emergence ) of RNA. Kim, E.-K.; Martin, V.; Krishnamurthy, R.Chem. Eur. J. 2017, 23, 12668-12675


- Investigations towards the synthesis of 5-amino-L-lyxofuranosides and 4-amino-lyxopyranosides and NMR analysis. Alba Diez-Martinez, A.; Krishnamurthy, R. SynOpen, 2017, 1, 29-40

- Anchimeric-assisted Spontaneous Hydrolysis of Cyanohydrins Under Ambient Conditions: Implications for Cyanide Initiated Selective Transformations. Yerabolu, J. R.; Liotta, C.L.; Krishnamurthy, R. Chem. Eur. J. 2017, 23, 8756-8765.


- Reaction of Glycine with Glyoxylate: Competing Transaminations, Aldol Reactions, and Decarboxylations. Conley, M.; Mojica, M.; Mohammed, F.; Chen, K.; Napoline , J. W.; Pollet, P.; Krishnamurthy, R.; Liotta, C. L. J. Phys. Org. Chem. 2017, 30, e3709.


- Giving Rise to Life: Transition from Prebiotic Chemistry to Protobiology. Krishnamurthy, R. Acc. Chem Res. 2017, 50, 455–459.

- Prebiotic Organic Chemistry and Chemical pre-Biology: Speaking to the Synthetic Organic Chemists. Krishnamurthy, R.; Snieckus, V. Synlett, 2017, 28, 27-29.


- Nucleobase Modification by an RNA Enzyme. Poudyal, R. R.; Ngyuyen, P. D. M.; Lokugamage, M. P.; Callaway, M. K.; Gavette, J. V.; Krishnamurthy, R.; Burke, D. H. Nucleic Acids Res. 2017, 45, 1345-1354.


-
A Plausible Prebiotic Origin of Glyoxylate: Nonenzymatic Transamination Reactions of Glycine with Formaldehyde. Mohammed, F. S.; Chen, K.; Mojica, M.; Conley, M.; Napoline, J. W.; Butch, C. J.; Pollet, P.; Krishnamurthy, R.; Liotta, C. L. Synlett, 2017, 28, 93-97.


-
Mineral-Induced Enantioenrichment of Tartaric Acid, Gherase, D.; Hazen, R. M.; Krishnamurthy, R.; Blackmond, D. Synlett, 2017, 28, 88-92.


- The Abiotic Oxidation of Organic Acids to Malonate.Rice, G. B.; Yerabolu, J. R.; Krishnamurthy, R.; Springsteen, G. Synlett, 2017, 28, 98-102.


- Kinetics of prebiotic depsipeptide formation from the ester–amide exchange reaction. Yu, S-S.; Krishnamurthy, R.; Fernandez, F.; Hud, N. V.; Schork, F. J.; Grover, M. A.Phys. Chem. Chem. Phys. 2016, 18, 28441-28450.

- RNA-DNA Chimeras in the Context of an RNA-world Transition to an RNA/DNA-world. Gavette, J. V.; Stoop. M.; Hud, N. V.; Krishnamurthy, R. Angew. Chemie, Int, Ed. 2016, 55, 13204-13209.

- Spontaneous Formation and Base Pairing of Plausible Prebiotic Nucleotides in Water. Cafferty, B. J.; Fialho, D.; Khanam, J.; Krishnamurthy, R.; Hud, N.Nature Communications 2016, 7, Article number: 11328.

- Small molecule-mediated duplex formation of nucleic acids with ‘incompatible’ backbones. Cafferty, B. J.; Musetti, C.; Kim, K.; Horowitz, E.D.; Krishnamurthy, R.; Hud, N. V. ChemComm. 2016, 52, 5436-5439.


- pH Controlled Reaction Divergence of Decarboxylation versus Fragmentation in Reactions of Dihydroxyfumarate with Glyoxylate and Formaldehyde: Parallels to Biological Pathways. Butch, C.J.; Wang, J.; Gu, J.; Vindas, R.; Crowe, J.; Pollet, P.; Gelbaum, L.; Leszczynski, J.; Krishnamurthy, R.; L. Liotta, C. L. J. Phys. Org. Chem. 2016, 29, 352-360.



- Hydrogen-Bonding Complexes of 5-Azauracil and Uracil Derivatives in Organic Medium. Diez-Martinez, A.; Kim, E-K.; Krishnamurthy, R. J. Org. Chem. 2015, 80, 7066-7075.

- Ester-Mediated Amide Bond Formation Driven by Wet-Dry Cycles: A Possible Path to Polypeptides on Prebiotic Earth. Forsythe, J.G.; Yu, S-S.; Mamajanov, I.; Grover, M.A.; Krishnamurthy, R.; Fernandez, F.M.; Hud, N.H. Angew. Chem. Int . Ed. 2015, 54, 9871-9875, DOI: 10.1002/ange.201503792.


- Synthesis of Orotidine by Intramolecular Nucleosidation. Kim, E-K.; Krishnamurthy, R. ChemComm. 2015, 51, 5618-5621.

- The Emergence of RNA. Krishnamurthy, R. Israel J. Chemistry, 2015, 55, 837-850; Cover page


- Microwave-Assisted Phosphitylations of DNA and RNA Nucleosides and Their Analogs. Efthymiou, T.; Krishnamurthy, R. Curr. Protoc. Nucleic Acid Chem. 60:2.19.1-2.19.20, 2015,DOI:10.1002/0471142700.nc0219s60.


- Synthesis of phosphoramidites of isoGNA, an isomer of glycerol nucleic acid. Kim, K.; Punna, V.; Karri, P.; Krishnamurthy, R.Beil. J. Org. Chem. 2014, 10, 2131-2138.


- A Plausible Simultaneous Synthesis of Amino Acids and Simple Peptides on the Primordial Earth. Parker, E. T.; Zhou, M.; Burton, A. S.; Glavin, D. P.; Dworkin, J. P.; Krishnamurthy, R.; Fernandez, F. M.; Bada, J. L. Angew. Chem. Int. Ed. 2014, 53, 8132-8136.Illustrated Back Cover.

- Microwave-Assisted Preparation of Nucleoside-Phosphoramidites. Meher, G.; Efthymiou, T.; Stoop, M.; Krishnamurthy, R. ChemComm, 2014, 50, 7463-7465.


-
RNA as an Emergent Entity: An Understanding Gained Through Studying its Non-Functional Alternatives. Krishnamurthy, R. Synlett, 2014, 25, 1511-1518.


- Spontaneous Prebiotic Formation of a β-Ribofuranoside That Self-Assembles with a Complementary Heterocycle. Chen, M.C.; Cafferty, B.J.; Mamajanov, I.; Gallego, I.; Khanam, J.; Krishnamurthy, R.; Hud, N. V. J. Am. Chem. Soc. 2014, 136, 5640-5646.

- Production of Tartrates by Cyanide Mediated Dimerization of Glyoxylate: A Potential Abiotic Pathway to the Citric Acid Cycle. Butch, C.; Cope, E.D.; Pollet, P.L.; Gelbaum, L.; Krishnamurthy, R.; Liotta, C. J. Am. Chem. Soc. 2013, 135, 13440-13445.

- Chemical Etiology of Nucleic Acid Structure. The Pentulofuranosyl Oligonucleotide Systems: (1'→3')-β-L-Ribulo, (4'→3')-α-L-Xylulo, and (1'→3')-α-L-Xylulo Nucleic Acids. Stoop, M.; Meher, G.; Karri, P.; Krishnamurthy, R. Chem. Eur. J. 2013, 19, 15336-15345.

- Base-Pairing Properties of a Structural Isomer of Glycerol Nucleic Acid. Karri, P.; Punna, V.; Kim, K.; Krishnamurthy, R. Angew. Chem. Int. Ed. 2013, 52, 5840-5844.

- The Origin of RNA and ‘‘My Grandfather’s Axe’’. Hud, N.; Cafferty, B.J.; Krishnamurthy, R.; Williams, L.D. Chemistry & Biology, 2013, 20, 466-474.
- Role of pKa of Nucleobases in the Origins of Chemical Evolution. Krishnamurthy, R. Acc. Chem. Res. 2012, 45, 2035-2044. Correction.

- A Unified Mechanism for Abiotic Adenine and Purine Synthesis in Formamide. Hudson, J. S.; Eberle, J. F.; Vachhani, R. H.; Rogers, L. C.; Wade, J. H.; Krishnamurthy, R.; Springsteen, G. Angew. Chemie. Int. Ed. 2012, 51, 5134-5137.

- Exploratory Experiments on the Chemistry of the "Glyoxylate Scenario": Formation of Ketosugars from Dihydroxyfumarate. Sagi, V.N.; Punna, V.; Hu, F.; Meher, G.; Krishnamurthy, R. J. Am. Chem. Soc. 2012, 134, 3577-3589. PMCID# PMC3284196

- Diastereoselective Self-Condensation of Dihydroxyfumaric Acid in Water: Potential Route to Sugars. Sagi, V.N.; Karri, P.; Hu, F., Krishnamurthy, R. Angew. Chem. Int. Ed. 2011, 50, 8127-8130.

- An expedient synthesis of L-ribulose and derivatives. Meher, G.; Krishnamurthy, R. Carbohydr. Res. 2011, 346, 703-707.

- Mapping the Landscape of Potentially Primordial Informational Oligomers: (3’→2’)-D-Phosphoglyceric Acid Linked Acyclic Oligonucleotides Tagged with 2,4-Disubstituted 5-Aminopyrimidines as Recognition Elements. Hernández-Rodríguez M.; Xie, J.; Osornio, Y. M.; Krishnamurthy, R. Chemistry An Asian Journal, 2011, 6, 1251-1262.


- Mapping the Landscape of Potentially Primordial Informational Oligomers: Oligo-Dipeptides Tagged with 6-Carboxy-pyrimidines as Recognition Elements. Zhang, X.; Krishnamurthy, R. Angew. Chem. Int. Ed. 2009, 48, 8124-8128.

- A search for Structural Alternatives of RNA. Krishnamurthy, R. J. Mex. Chem. Soc. 2009, 53, 23-33.

- Structure of TNA-TNA complex in solution: NMR Study of the Octamer Duplex Derived from α-(L)-threofuranosyl-(3’–2’)-CGAATTCG. Ebert, M-O.; Mang, C.; Krishnamurthy, R.; Eschenmoser, A.; Jaun, B. J. Am. Chem. Soc. 2008, 130, 15105-15115.

- Mapping the Landscape of Potentially Primordial Informational Oligomers: Oligo-Dipeptides Tagged with 2,4-Disubstituted 5-amino-pyrimidines as Recognition Elements. Mittapalli, G.K.; Osornio, Y.M.; Guerrero, M.A.; Ravinder, K.R.; Krishnamurthy, R.; Eschenmoser, A. Angew. Chem. Int. Ed. 2007, 46, 2478-2484.

- Mapping the Landscape of Potentially Primordial Informational Oligomers: Oligo-dipeptides and Oligo-dipeptoids Tagged with Triazines as Recognition Elements. Mittapalli, G.K.; Ravinder, K.R.; Xiong, H.; Munoz, O.; Han, B.; De Riccardis, De F.; Krishnamurthy, R.; Eschenmoser, A. Angew. Chem. Int. Ed. 2007, 46, 2470-2477.

- Tautomerism in 5,8-Diaza-7,9-dicarbaguanine (‘Alloguanine’). Wagner, T.; Han, B.; Krishnamurthy, R.; Eschenmoser, A. Helv. Chim. Acta. 2005, 88, 1960-1968.
- Mannich-Type C-nucleosidations with 7-Carba-purines and 4-Amino-pyrimidines. Han, B.; Rajwanshi, V.; Nandy, J.; Krishnamurthy, R.; Eschenmoser, A. Synlett. 2005, 744-750.
- Mannich-Type C-Nucleosidations in the 5,8-Diaza-7,9-dicarba-purine Family. Han, B.; Jaun, B.; Krishnamurthy, R.; Eschenmoser, A. Org. Lett. 2004, 6, 3691-3694.
- Base-Pairing Systems Related to TNA Containing Phosphoramidate Linkages: Synthesis of Building Blocks and Pairing Properties. Ferenic, M.; Reddy, G.; Wu, X.; Guntha, S.; Nandy, J.; Krishnamurthy, R.; Eschenmoser, A. Chemistry & Biodiversity, 2004, 1, 939-979.
- The β-D-Ribopyranosyl-(4’→2’)-oligonucleotide System (‘pyranosyl-RNA’): Synthesis and Resumé of Base-Pairing Properties. Pitsch, S.; Wendeborn, S.; Krishnamurthy, R.; Holzner, A.; Minton, M.; Bolli, M.; Miculca, C.; Windhab, N.; Micura, R.; Stanek, M.; Jaun, B.; Eschenmoser, A. Helv. Chim. Acta. 2003, 86, 4270-4363.
- Assignment of the 1H and 13C-NMR Spectra of N2,N6-dibenzoyl-N2,N9-bis(2’,3’-di-O-benzoyl-(a)-L-Threofuranosyl)-2,6-diaminopurine. Delgado, G.; Krishnamurthy, R. Revista de la Sociedad Quimica de Mexico, 2003, 47, 216-220.
- Why Does TNA Cross-Pair More Strongly with RNA Than with DNA? An Answer From X-ray Analysis. Pallan, P. S.; Wilds, C. J.; Wawrzak, Z.; Krishnamurthy, R.; Eschenmoser, A., Egli., M Angew. Chem. Int. Ed. 2003, 42, 5893-5895.
- C-Nucleosidations with 2,6-Diamino-5,8-diaza-7,9-dicarba-purine. Han, B.; Wang, Z.; Jaun, B.; Krishnamurthy, R.; Eschenmoser, A. Org. Lett. 2003, 5, 2071-2074.
- 2,6-Diamino-5,8-diaza-7,9-dicarba-purine. Wang, Z.; Huynh, H. K.; Han, B.; Krishnamurthy, R.; Eschenmoser, A. Org. Lett. 2003, 5, 2067-2070.
- Pentopyranosyl Oligonucleotide Systems. The α-L-Arabinopyranosyl-(4’→2’)-Oligonucleotide System: Synthesis and Pairing Properties. Jungmann, O.; Beier, M.; Luther, A.; Huynh, H. K.; Ebert, M. O.; Jaun, B.; Krishnamurthy, R.; Eschenmoser, A. Helv. Chim. Acta. 2003, 86, 1259-1308.
- The α-L-Threofuranosyl-(3’→2’)-Oligonucleotide System (‘TNA’): Synthesis and Pairing Properties. Schoning, K.-U.; Scholz, P.; Wu, X.; Guntha, S., Delgado, G.; Krishnamurthy, R.; Eschenmoser, A. Helv. Chim. Acta. 2002, 85, 4111-4153.
- Crystal Structure of a B-Form DNA Duplex Containing L-α-Threofuranosyl-(3’→2’)-Nucleosides: A Four-Carbon sugar is easily accommodated into the back bone of DNA. Wilds, C. J.; Wawrzak, Z.; Krishnamurthy, R.; Eschenmoser, A.; Egli, M. J. Am. Chem. Soc. 2002, 124, 13716-13721.
- NMR Solution Structure of Duplex Formed by Self-Pairing of α-(D)-Arabinopyranosyl-(4’→2’)-(CGAATTCG). Ebert, M-O.; Hoan, H. K.; Luther, A.; Krishnamurthy, R.; Eschenmoser, A., Jaun, B. Helv. Chim. Acta. 2002, 85, 4055-4073.
- 2,6-Diaminopurines in TNA: Effect on Duplex Stabilities and on the Efficiency of Template-Controlled Ligations. Wu, X.; Delgado, G.; Krishnamurthy, R.; Eschenmoser, A. Org. Lett. 2002, 4, 1283-1286.
- Base-Pairing Systems Related to TNA: α-Threofuranosyl Oligonucleotides Containing Phosphoramidate Linkages. Wu, X.; Guntha, S.; Ferencic, M.; Krishnamurthy, R.; Eschenmoser, A. Org. Lett. 2002, 4, 1279-1282.
- Pentopyranosyl Oligonucleotide Systems. β-(D)-Xylopyranosyl-(4’→2’)-oligonucleotide System. Wagner, T.; Hoan, H. K.; Krishnamurthy, R.; Eschenmoser, A. Helv. Chim. Acta. 2002, 85, 399-416.
- Pentopyranosyl Oligonucleotide Systems. Systems with Shortened Backbones: (D)-β-Ribopyranosyl-(4’→3’)- and (L)-α-Lyxopyranosyl-(4’→3’)-oligonucleotide System. Wippo, H.; Reck, F.; Kudick, R.; Ramasehsan, M.; Ceulemans, G., Bolli, M.; Krishnamurthy, R.; Eschenmoser, A. Bioorg. Med. Chem. 2001, 9, 2411-2428.
- Pentopyranosyl Oligonucleotide Systems. The α-L-Lyxopyranosyl-(4’→2’)-oligonucleotide System. Reck, F.; Wippo, H.; Kudick, R.; Krishnamurthy, R.; Eschenmoser, A. Helv. Chim. Acta. 2001, 84, 1778-1804.
- Chemical Etiology of Nucleic Acid Structure: The α-Threofuranosyl-(3’→2’) Oligonucleotide System. Schöning, K.-U.; Scholz, P.; Guntha, S.; Wu, X.; Krishnamurthy, R.; Eschenmoser, A. Science 2000, 290, 1347-1351.
- Concentration of Simple Aldehydes by Sulfite-Containing Double-Layer Hydroxide Minerals: Implications for Biopoesis. Pitsch, S.; Krishnamurthy, R.; Arrhenius. G. Helv. Chim. Acta. 2000, 83, 2398.
- Regioselective a-Phosphorylation of Aldoses in Aqueous Solution. Krishnamurthy, R.; Guntha, S.; Eschenmoser. A. Angewandte Chemie Int. Ed. 2000, 39, 2281.
- Before RNA and After: Geophysical and Geochemical Constraints on Molecular Evolution. Mojzsis, S.; Krishnamurthy, R.; Arrhenius, G. in ‘The RNA World’, second edition, pp 1-47, Eds. Gesteland, R. F.; Cech, T. R.; Atkins, J. F. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York, 1999. DOI: 10.1101/087969589.37.1
- L-α-Lyxopyranosyl (4'→3') Oligonucleotides: A Base-Pairing System Containing a Shortened Backbone. Reck, F.; Wippo, H.; Kudick, R.; Krishnamurthy, R.; Eschenmoser, A. Organic Letters, 1999, 1, 1531-1534.
- Promiscuous Watson-Crick Cross-Pairing within the Family of Pentopyranosyl (4'→2') Oligonucleotides. Jungmann, O.; Wippo, H.; Stanek, M.; Huynh, H. K.; Krishnamurthy, R.; Eschenmoser, A. Organic Letters, 1999, 1, 1527-1530.
- Chemical Etiology of Nucleic Acid Structure: Comparing Pentopyranosyl-(2'→4') Oligonucleotides with RNA. Beier, M.; Reck, F.; Wagner, T.; Krishnamurthy, R.; Eschenmoser, A. Science 1999, 283, 699-703.
- Formation of Glycolaldehyde Phosphate From Glycolaldehyde in Aqueous Solution. Krishnamurthy, R.; Arrhenius, G; Eschenmoser, A. Origins Life Evol. Biosphere 1999, 29, 333-354.
- Mineral Induced Formation of Pentose-2,4-diphosphates. Krishnamurthy, R.; Pitsch, S.; Arrhenius, G. Origins Life Evol. Biosphere 1999, 29, 139-152.
- Formation of sugar phosphates under potentially natural conditions. Krishnamurthy, R.; Pitsch, S.; Eschenmoser, A.; Arrhenius, G. Mineral. Mag. 1998, 62A(Pt. 2), 815.
- Pyranosyl-RNA: Base-pairing beween Homochiral Oligonucelotide Strands of Opposite Sense of Chirality. Krishnamurthy, R.; Pitsch, S.; Minton, M.; Miculka, C.; Windhab, N.; Eschenmoser, A. Angewandte Chemie, Int. Ed. Engl. 1996, 35, 1537-1541.
- p-RNA, the pyranosyl isomer of RNA: Pairing properties and potential to replicate. Pitsch, S.; Krishnamurthy, R.; Wendeborn, S.; Holzner, A.; Minton, M.; Lesueur, C.; Schlönvogt, I.; Jaun, B.; Eschenmoser, A. Helevetica chimica Acta 1995, 78, 1621-1635.
- Bis(tri-n-butylstannyl)benzopinacolate: Preparation and Use as a Mediator of Intermolecular Free Radical Reactions. Hart, D. J.; Krishnamurthy, R.; PooK, L. M.; Seely, F. L. Tetrahedron Letters 1993, 34, 7819-7822.
- Synthesis of 6H-Dibenzo(b,d)pyran-6-ones via Dienone-Phenol Rearrangements of Spiro(2,5-Cyclohexadiene-1,1'(3'H)-isobenzofuran)-3'-ones. Hart, D. J.; Kim, A.; Krishnamurthy, R.; Merriman, G. H.; Waltos, A-M. Tetrahedron 1992, 48, 8179-8188.
- Investigation of a Model for 1,2-Asymmetric Induction in Reactions of a-Carbalkoxy Radicals: A Stereochemical Comparison of Reactions of α-Carbalkoxy Radicals and Ester Enolates. Hart, D. J.; Krishnamurthy, R. J. Org. Chem. 1992, 57, 4457-4470.
- Stereoselective Free Radical Reactions at C(20) of Steroid Chains. Hart, D. J.; Krishnamurthy, R., Synlett. 1991, 412-414.
- Free-Radical Cyclizations: Application to the Total Synthesis of dl-Pleuorotin and dl-Pleurotinic acid. Hart, D. J.; Huang, H.-C; Krishnamurthy, R.; Schwartz, T. J. Am. Chem. Soc. 1989, 111, 7507-7519.
California: 10550 North Torrey Pines Road, La Jolla, CA 92037 - (858) 784-1000
Florida: 130 Scripps Way, Jupiter, FL 33458 - (561) 228-2000
Copyright © 2025, The Scripps Research Institute (TSRI). All Rights Reserved.