

Neurons grown in culture expressing a mutant prion protein (cyan) that cause prion disease in humans. These neurons display swollen axons that contain toxic mutant prion protein aggregates. Chassefeyre et al. identified genes that account for the formation of these aggregates and showed that reducing their function can inhibit aggregate formation and prevent neuronal dysfunction. (Image Credit: Adriaan Verhelle and Yin Wu, Scripps Research.)
Scripps Research discovery illuminates how brain cells die in prion diseases
Researchers show how toxic aggregates are formed inside brain cells, and how to block the cell-killing process—which may also be at work in Alzheimer’s and other neurodegenerative diseases.
December 22, 2021
LA JOLLA, CA—Prion diseases, such as Creutzfeldt-Jakob Disease (CJD), are fast-moving, fatal dementia syndromes associated with the formation of aggregates of the prion protein, PrP. How these aggregates form within and kill brain cells has never been fully understood, but a new study from scientists at Scripps Research suggests that the aggregates kill neurons by damaging their axons, the narrow nerve fibers through which they send signals to other neurons.
The accumulation of protein aggregates in axons, along with axonal swellings and other signs of dysfunction, are also early features of other neurodegenerative disorders including Alzheimer’s and Parkinson’s diseases. The discovery of how these prion aggregates form in axons and how to inhibit them, reported in Science Advances, may ultimately have a significance that goes far beyond prion diseases.
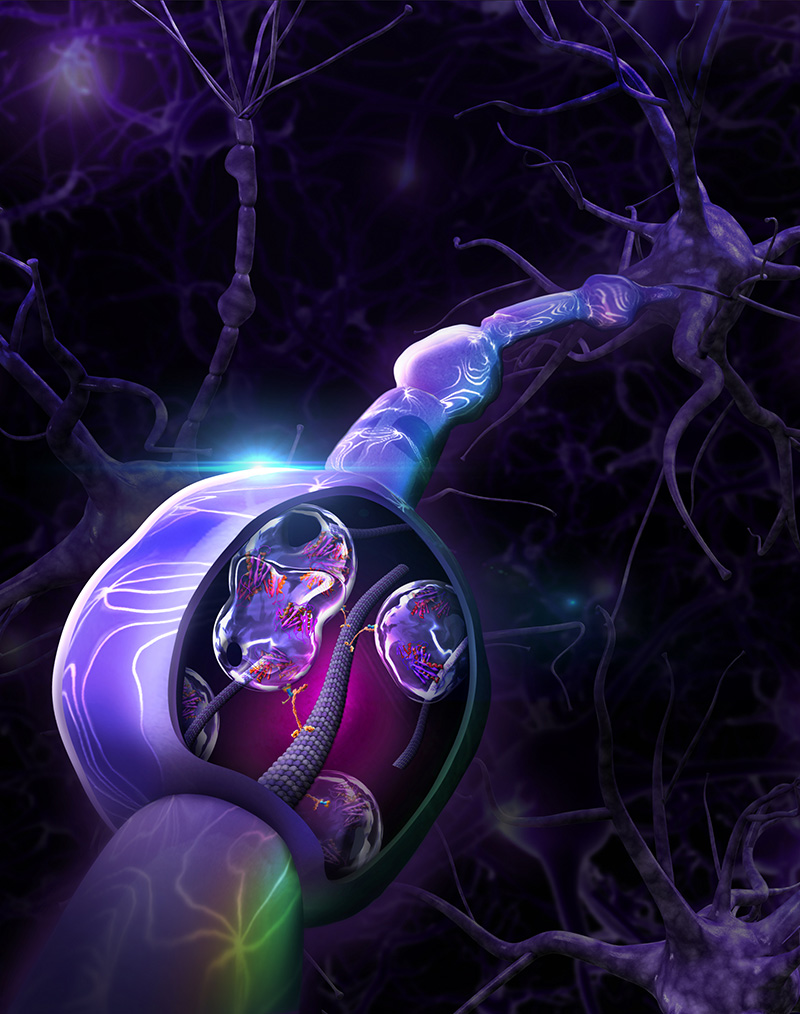
This image, which appears as the Dec 22, 2021 Science Advances cover, shows a rendering of large aggregates that form in neuronal axons in cases of prion diseases. The Encalada Lab at Scripps Research has shown how these aggregates form and how to inhibit them, paving the way for possible new treatments. (Image Credit: Rob Heller.)
“We’re hopeful that these findings will lead to a better understanding of prion and other neurodegenerative diseases, as well as new strategies for treating them,” says study senior author Sandra Encalada, PhD, Arlene and Arnold Goldstein Associate Professor in the Department of Molecular Medicine at Scripps Research.
The researchers in their study closely observed mutant, disease-causing copies of the prion-disease protein PrP forming large aggregates in the axons of neurons, but not in the neurons’ main cell bodies. The formation of these aggregates was followed by signs of axon dysfunction and ultimately neuronal death. The scientists found evidence that neurons’ waste-disposal processes normally are able to cope with such aggregates when they are within or close to neurons’ main cell bodies, but are much less able to do so when the aggregates accumulate far out within axons.
The researchers also identified a complex of key proteins as being responsible for steering PrP into axons and causing aggregation associated with large axonal swellings. They demonstrated that by silencing any one of these proteins they could inhibit the aggregates from forming and protect the neurons from damage and death.
Vulnerable axons
CJD is the most common human prion disease, occurring at the rate of about one case per million people per year worldwide. Most cases are thought to arise spontaneously when PrP somehow is altered in the brain and starts aggregating. Because these aggregates grow by a chain-reaction process that draws in healthy copies of PrP, they can transmit CJD in rare cases —for example, during corneal transplant surgery—from one person to another. About 15 percent of cases are hereditary, caused by mutations that make PrP more likely to aggregate. Prion disorders occur in other mammals and are thought to be due to similar toxic aggregations of different species’ PrP proteins.
In the study, Encalada’s team used mouse brain cells containing mutant PrP, along with microscopic motion-picture techniques, to study the initial accumulation of PrP aggregates in axons. A neuron’s axon is often very long in relation to its main body—the soma—and has been found to be uniquely vulnerable to disruptions of its delicate systems for transporting essential molecules and getting rid of waste.
PrP’s ordinary function in neurons has never been clear, but the protein appears to be normally secreted, via sac-like containers called vesicles, from the soma and the axon, where it sometimes returns to be recycled or degraded as waste. The researchers found in their experiments that mutant PrP produced in the soma is also largely encapsulated in vesicles that get moved into the axon along railways called microtubules.
This movement involves a somewhat complex vesicle trafficking system, and the researchers observed that this system shunts much of the PrP far out into axons, where PrP-containing vesicles gather and merge. Mutant PrP in this situation forms large aggregates—Encalada calls them endoggresomes—that axons can’t get rid of. The aggregates lead to axonal swellings, and other signs of dysfunction including reduced neuronal calcium signaling, and ultimately a much faster neuronal death rate compared to neurons with normal PrP.
The researchers also found a way of countering endoggresomes formation. They identified four proteins, Arl8, kinesin-1, Vps41, and SKIP, that are responsible for directing PrP-containing vesicles into axons, carrying them far out into the soma, and merging them with other PrP-containing vesicles to trigger aggregate formation. When they silenced any of these proteins, far fewer PrP-containing vesicles entered axons, the axons showed few or no signs of aggregation, and the neurons functioned normally or almost normally and survived just as well as normal brain cells.
The results point to the tantalizing possibility that prion diseases, and perhaps many other protein-aggregate diseases of the brain, can be prevented or treated by interrupting at least transiently the trafficking process that brings vesicle-encapsulated, aggregate-prone proteins out into axons.
“We’re very enthusiastic about discovering molecules that can inhibit this aggregate-forming pathway and studying the effects of such inhibitors in animal models of prion and other neurodegenerative diseases,” Encalada says.
“Endosomal Sorting Drives the Formation of Axonal Prion Protein Endoggresomes” was co-authored by Romain Chassefeyre, Tai Chaiamarit, Adriaan Verhelle, André Leitão and Sandra Encalada, all of Scripps Research; and Sammy Weiser Novak, Leonardo Andrade and Uri Manor, of the Salk Institute for Biological Studies.
The research was funded by the National Institutes of Health (R01AG049483) and others.
For more information, contact press@scripps.edu